Block Copolymer Solar Cells
Bulk heterojunctions in plastic solar cells

Polymer-based organic photovoltaics are most commonly made from blends of a conjugated polymer and fullerene, the electron donor and acceptor, respectively. Light induces a charged state, known as an exciton, in which the electron has the potential to separate from its hole (the hole is the empty atomic orbital, conventionally viewed as a positive charge) and flow as electricity. In order for charge separation to occur, the exciton must diffuse to the interface between the donor and acceptor. Bulk heterojunctions are highly dispersed mixtures of the electron donor and acceptor, yielding a large interface for charge separation. The greatest efficiencies are expected when the characteristic length of the bulk heterojunction is ~10 nm because that is the diffusion length of an exciton. Fig. 1 shows both bulk heterojunctions with both disordered and ordered morphologies.
Creating nanonstructured bulk heterojunctions is only half of the challenge, preventing them from coarsening is a separate problem. The phase separated donor and acceptor structures tend to coarsen in order to decrease the free energy of their interface, resulting in less interfacial area for the bulk heterojunction. Block copolymers address both challenges by self-assembling into ordered nanostructures that are thermodynamically equilibrated, and therefore stable against coarsening. Early attempts to use block copolymers in solar cells, nicely summarized in a review by Paul Topham et al.1, have uncovered a complex interplay of thermodynamic and kinetic forces. Continuing research and creative processing are needed to realize the potential of block copolymer-based solar cells.
All-conjugated diblock copolymers in solar cells
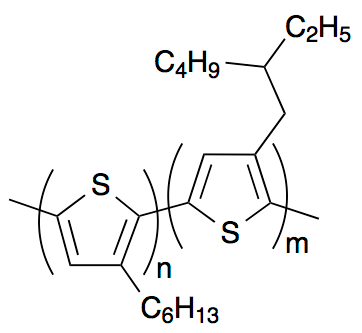
In recent work, we designed an all-conjugated diblock copolymer that was blended with the fullerene phenyl-C61-butyric acid methyl ester (PCBM) to produce solar cells. The rationale for choosing a donor-block-donor, rather than a donor-block-acceptor, was that solar cell efficiency is limited by the total amount of donor phase available to produce excitons. The diblock copolymer, shown in Fig. 2, comprised poly-3-hexylthiophene (P3HT) covalently bonded to poly-3-ethyl-hexyl-thiophene (P3EHT). P3HT is semi-crystalline and P3EHT is fully amorphous. The diblock copolymer was symmetric, and self-assembled to form alternating crystalline and amorphous lamellae, as shown in Fig. 3. PCBM dissolved into the amorphous regions of the diblock copolymer, precipitating at concentrations above the miscibility limit.

The size of nanostructured domains, in this case lamellae, is strongly correlated with the molecular weight of the diblock copolymer. Lower molecular weights result in smaller structures, which is desirable for bulk heterojunctions. Higher molecular weights (parameterized in polymer physics by χN), however, are desirable for driving the diblock copolymer to self-assemble. In the case of P3HT-block-P3EHT, the two blocks are so chemically similar that the copolymer does not spontaneously self-assemble by traditional nanophase separation. Rather, the block copolymer self assembles because the P3HT block crystallizes.Polymer crystallinity generally increases with decreasing molecular weight, because short chains can assemble into a crystalline structure more easily than long entangled chains. Selecting a low molecular weight diblock copolymer (11 kg mol-1) enabled us to simultaneously decrease the size of bulk heterojunctions and increase the driving force for self-assembly.
Fullerene loading and self-assembly
We varied the concentration of PCBM in the diblock from 0 – 65 wt%, examining the morphology and crystallinity of the resulting thin films. Processing, of course, plays a large role in the development of nanostructure. These films were ~100 nm thick, coated by a wirebar, dried under ambient conditions, then under vacuum, annealed at 200 ºC (around the melting temperature of the P3HT block) for 6 hours, and finally cooled slowly to room temperature. The crystallinity of the polymer increased linearly with PCBM loading, even in films below the miscibility limit of PCBM (between 10 and 35 wt%), indicating cooperativity between P3HT crystallization and segregation of PCBM into the amorphous P3EHT block. The top surface nanostructures also changed with PCBM loading, exhibiting an optimum in lamellar ordering for PCBM concentrations on the range of 10 – 40 wt%. We suspect, based on strong grazing incidence x-ray diffraction in the plane of incidence, that the crystalline lamellar structures are propagating through the film.
A nucleating agent to reduce the size of fullerene crystals

The most obvious effect of PCBM was on the microscopic structure of the film. The thin films dewetted with increased PCBM loading, due to the low molecular weight of the copolymer and low density of crystalline domains to serve as cross-links. The growth of microscopic PCBM crystals drove capillary instabilities which ruptured the film. Nucleating agents resolve this problem by creating a very large population of small PCBM crystals (as opposed to a small population of large crystals). We selected a low molecular weight homopolymer, P3HT Mw = 2 kg mol-1, as the nucleating agent, to avoid using inactive filler. The P3HT nucleating agent had the desired effect of preserving a smooth film surface without any measurable impact upon the lamellar nanostructure. The use of such ternary blends is promising because it delivers structural control whilst reducing the total amount of block copolymer needed. Given that the polyethylene terephthalate lamination used to encase plastic solar cells is the greatest material cost3, implementing the use of more expensive copolymers is economically feasible.
References
[3] Cara J. Mulligan, Mitchell Wilson, Glenn Bryant, Ben Vaughan, Xiaojing Zhou, Warwick J. Belcher, and Paul C. Dastoor. A projection of commercial-scale organic photovoltaic module costs. Solar Energy Materials and Solar Cells, 120:9-17, 2014.